BAD Day 1: Multiple hypothesis testing on Golub
In this tutorial we will do multiple hypothesis testing on the previsouly
studied Golub data set.
- Null Hypothesis $H_0$: states the assumption to be tested. We begin by assuming that the null Hypothesis is True
- Alternative Hypothesis $H_1$: is the opposite of the null hypothesis
1. Loading the dataset
require(multtest)
data(golub)Gene expression data (3051 genes and 38 tumor mRNA samples) from the leukemia microarray study of Golub et al. (1999). Pre-processing was done as described in Dudoit et al. (2002). The R code for pre-processing is available in the file ../doc/golub.R.
golub.expr<-golub
row.names(golub.expr)=golub.gnames[,3]Numeric vector indicating the tumor class, 27 acute lymphoblastic leukemia (ALL) cases (code 0) and 11 acute myeloid leukemia (AML) cases (code 1).
colnames(golub.expr)= golub.clSetting the sample sizes
n.ALL <- 27
n.AML <- 11
cancer.type <- c(rep("ALL", n.ALL), rep("AML", n.AML))Adding the cancer type to the column name, for the display
colnames(golub.expr) <-cancer.type2. t.test with a single gene
g <- 347Alternatively, you can select a gene randomly
## g <- sample(1:nrow(golub.expr),1)
g.profile <- as.vector(as.matrix(golub.expr[g,]))Draw a barplot wiht color-coded cancer type
plot.col <- c('ALL'='mediumpurple', 'AML'='midnightblue')
barplot(g.profile, main = paste("Golub (1999), gene", g),
col = plot.col[cancer.type], border = 'white')
legend('topright', c("ALL","AML"),col = plot.col[c("ALL","AML")],
pch = 15, bty = "o")
box()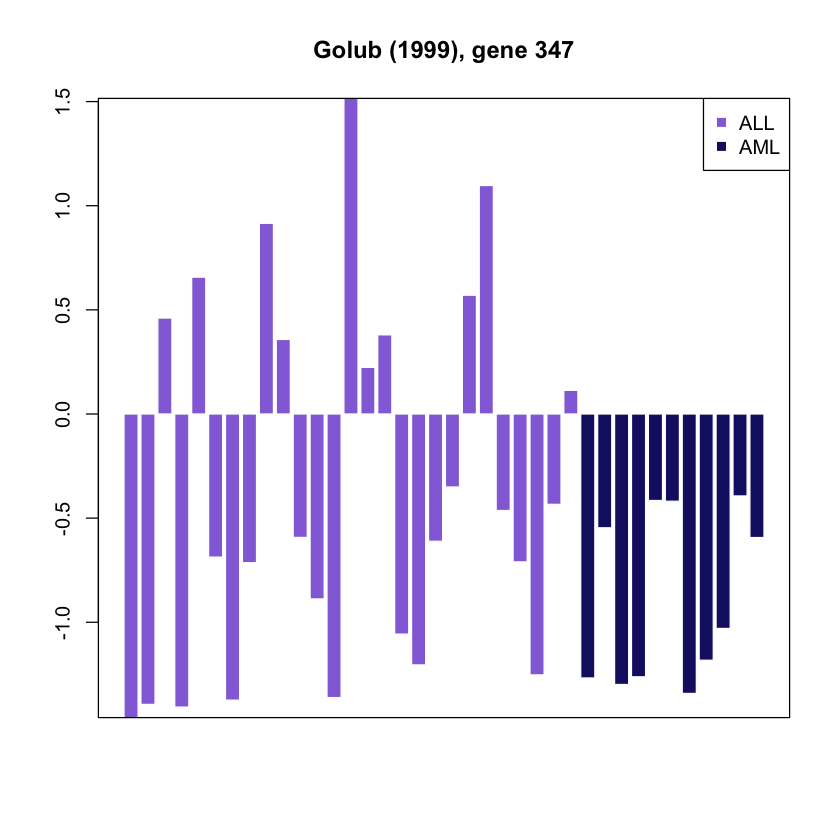
Separate data in two vectors
sample.ALL <- g.profile[cancer.type=="ALL"]
sample.AML <- g.profile[cancer.type=="AML"]Compute manually the t test paramenters (not necessary, just to practice!)
Estimate the population means
mean.est.ALL <- mean(sample.ALL)
mean.est.AML <- mean(sample.AML)Compute the sample sd
Remember the sd( ) function automatically computes the estimate corrected with
sqrt(n-1)
sample.sd.ALL <- sd(sample.ALL) * sqrt((n.ALL-1)/n.ALL)
sample.sd.AML <- sd(sample.AML) * sqrt((n.AML-1)/n.AML)Estimate the population standard deviation
sd.est.ALL <- sd(sample.ALL)
sd.est.AML <- sd(sample.AML)Estimate the standard errors on the mean
sd.err.est.ALL <- sd(sample.ALL) / sqrt(n.ALL)
sd.err.est.AML <- sd(sample.AML) / sqrt(n.AML)Estimate the standard deviation of the difference between the two means according to Student’s formula
diff.sd.est <- sqrt((n.ALL*sample.sd.ALL^2 + n.AML*sample.sd.AML^2) * (1/n.ALL + 1/n.AML) /(n.ALL+n.AML-2))Compute t.obs
d <- abs(mean.est.ALL - mean.est.AML)
t.obs.Student <- d/diff.sd.estCompute the P- vale. Since we are performing the two-tail test, the single-tail probability has to be multiplied by 3 in order to obtai the alpha risk
P.val.Student <- 2 * pt(q = t.obs.Student, df = n.ALL + n.AML-2, lower.tail = F)This is what you should be doing… FAST!
3. Apply the Student-Fischer t-test
Note: this assumes that the two populations have equal variance.
t.student <- t.test(sample.ALL,sample.AML, var.equal=TRUE)
print(t.student)4. Apply the Welch t-test
Note: this does not assume that the two populations have equal variance.
t.welch <- t.test(sample.ALL,sample.AML, var.equal=FALSE)
print(t.welch)We want to test all the genes, so we could just loop over what was written before … (note it can take some time)
t.statistics <- vector()
P.values <- vector()
for (g in 1:nrow(golub.expr)) {
# print(paste("Testing gene", g))
g.profile <- as.vector(golub.expr[g,])
sample.ALL <- g.profile[cancer.type=="ALL"]
sample.AML <- g.profile[cancer.type=="AML"]
t <- t.test(sample.ALL,sample.AML)
t.statistics <- append(t.statistics, t$statistic)
P.values <- append(P.values, t$p.value)
} print(P.values)For a more efficient way, you can use apply instead (not shown here)
Data=cbind(golub.expr,P.values)
colnames(Data)[39]='Raw.p'Order data by pvalue
Data = Data[order(Data[,'Raw.p']),]Perform p-value adjustments and add to the data frame
Datadf <- as.data.frame(Data)Datadf$Bonferroni =
p.adjust(Datadf$Raw.p,
method = "bonferroni")
Datadf$BH =
p.adjust(Datadf$Raw.p,
method = "BH")
Datadf$Holm =
p.adjust(Datadf$ Raw.p,
method = "holm")
Datadf$Hochberg =
p.adjust(Datadf$ Raw.p,
method = "hochberg")
Datadf$Hommel =
p.adjust(Datadf$ Raw.p,
method = "hommel")
Datadf$BY =
p.adjust(Datadf$ Raw.p,
method = "BY")5. Plotting the obtained data
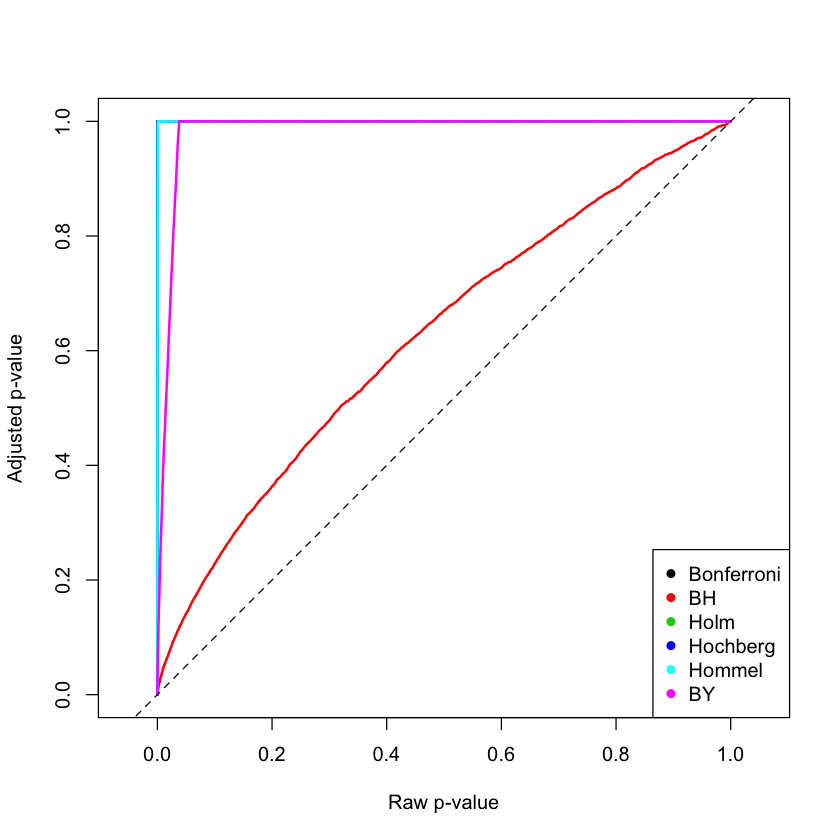
X = Datadf$Raw.p
Y = cbind(Datadf$Bonferroni,
Datadf$BH,
Datadf$Holm,
Datadf$Hochberg,
Datadf$Hommel,
Datadf$BY)matplot(X, Y,
xlab="Raw p-value",
ylab="Adjusted p-value",
type="l",
asp=1,
col=1:6,
lty=1,
lwd=2)
legend('bottomright',
legend = c("Bonferroni", "BH", "Holm", "Hochberg", "Hommel", "BY"),
col = 1:6,
cex = 1,
pch = 16)
abline(0, 1,
col=1,
lty=2,
lwd=1)